English Translation by: Ting Wu, Hao Zhu, Meng Su
Disclaimer: The English is for information only and not an official translation and under any dispute the Chinese will prevail
Center for Drug Evaluation, NMPA
November 2024
Table of Contents
iii............ Generating Clinical Evidence as an External Control for Single-Arm Trials........ 5
V........... Evaluation of Disease Registry Data from a Clinical Research Perspective............ 15
VI......... Submission of Disease Registry Data and Communication with Regulatory Authorities 17
Guidance on the Application of Real-World Data Based on Disease Registries (Trial Version)
I. Introduction
The use of real-world evidence (RWE) to support regulatory decision-making in drug development is receiving increasing attention. However, a key challenge in conducting real-world studies (RWS) is that real-world data (RWD) often fall short—particularly in terms of quality, quantity, and accessibility—of meeting the clinical evidence standards required for regulatory purposes. Disease registries, which are typically designed prospectively with specific objectives, collect data on diseases and related clinical characteristics in defined populations and emphasize quality control throughout implementation. As a result, they generally offer higher data quality and serve as an important source of RWD. To generate robust RWE that can support regulatory decision-making, it is essential to fully utilize existing high-quality data resources and strategically develop well-designed disease registries.
This guidance outlines how to establish disease registries, as well as how to evaluate and utilize existing disease registry data, with the aim of providing guidance on the application of real-world data based on disease registries.
II. Definition of Disease Registries
The English term "registry" can be translated into Chinese as "注册" or "注册登记," among others. To distinguish it from "drug registration" in the context of clinical research, this guidance uses the term “登记” (registry).
A registry is a database created through the organized, systematic, and standardized collection of longitudinal, patient-level data related to demographics, diseases, exposures, diagnoses, treatments, and outcomes. This data collection is conducted based on predefined objectives, as well as specified follow-up or observation periods and time points. While registry data are typically collected prospectively, they may also incorporate historical data in accordance with the study design.
Registries collect patient-level data, including information on disease status, treatments received, and exposures (such as medication use). Depending on the focus of their content, registries can be classified into Disease Registry, Product Registry, and Health Service Registries. Disease registries may target a single disease (e.g., breast cancer registry, hypertension registry, Gaucher disease registry), a disease category (e.g., cancer registries, cardiovascular disease registries), or cover one or more disease types at a national or regional level (e.g., the China National Rare Disease Registry System). Given the multiple interpretations of the term “registry” and the practical needs of drug development and evaluation, this guidance primarily focuses on single-disease registries, unless otherwise specified. Other types of disease registries may refer to this guidance as applicable.
The greatest advantage of registry data lies in its ability to prospectively establish cohorts of patients with specific diseases based on clearly defined research objectives. This enables a targeted approach to determining what data should be collected and how, resulting in the development of comprehensive, longitudinal datasets. These datasets may include detailed information on medication use and the collection of patient experience data (PED), reflecting the principles of patient-focused drug development (PFDD)—such as patient-reported outcomes (PROs). Prospectively designed registries also support the creation of structured and standardized databases, while taking advantage of increasingly sophisticated digital tools to enable efficient and high-quality data capture.
Studies conducted using data generated from registries are referred to as registry-based studies.
If a registry-based study is poorly designed or inadequately implemented, the value of the registry data can be significantly compromised—primarily due to the introduction of various forms of bias. For example: Omission of key variables or lack of strict specifications during the design phase (e.g., measurement methods, follow-up intervals, and time points) can lead to missing or unusable critical information, resulting in various biases or hindering the correction of certain biases; inappropriate inclusion/exclusion criteria may affect the representativeness of the target population or introduce selection bias; patients with more severe conditions may be more likely to be enrolled than those with milder conditions, limiting the representativeness of the study population; high loss-to-follow-up rates may introduce additional selection bias; lack of sustained quality control can compromise data quality; data heterogeneity across study sites (e.g., differences in clinical practices, treatment standards, socioeconomic backgrounds, or self-management of disease capabilities) can introduce bias and pose challenges to both study design and interpretation of results.
III. Application Scenarios of Disease Registry Data
Registry-based studies have broad applicability, including use in understanding the natural history of diseases, identifying prognostic factors, characterizing diagnostic and treatment practices, monitoring safety risks, and assessing treatment effectiveness. In the context of drug development, disease registry data collected from real-world clinical settings can be utilized for both pre-marketing and post-marketing evaluations. Common application scenarios include, but are not limited to, the following:
i. Generating Primary or Supportive Clinical Evidence
Real-world evidence derived from disease registry data can serve as primary or supportive clinical evidence to inform regulatory decision-making. For example, data collected through registries in areas such as pediatrics, rare diseases, or oncology may be used to support regulatory decisions for new indication approvals.
ii. Providing a Basis for Clinical Trial Design
Using registry data can provide a certain basis for clinical trial design. It can support the development of inclusion and exclusion criteria, inform the selection of study endpoints that reflect clinical relevance and are sensitive to clinical efficacy, and guide the determination of key observation time points, time windows, and follow-up intervals. Registry data can also facilitate the exploration of the minimal clinically important difference (MCID) and help characterize patient healthcare-seeking behaviors and treatment preferences. In addition, if the registry includes comprehensive data on relevant genes or biomarkers, it can significantly enhance the precision of target population identification for clinical trials.
iii. Generating Clinical Evidence as an External Control for Single-Arm Trials
Single-arm study designs typically require the use of an external control. Forms of external controls include historical controls and concurrent controls, etc., and the source and selection of such controls are critical. External controls impose high requirements on the quality and sample size of real-world data. The baseline characteristics and key outcome-related variables (excluding the treatment under investigation) should closely resemble those of the trial group, and missing data should be kept within acceptable range. If there are well-designed and well-executed high-quality disease registry data, they should be prioritized as external controls because they are more likely to meet the requirements of external controls compared to other data sources and are more feasible in terms of data acquisition and curation.
iv. Studying the Natural History of Disease
The natural history of diseases is very important in clinical research, especially for rare diseases. Disease registry systems are commonly used platforms for obtaining such data. When well-designed, registries can provide valuable support for rare disease research by capturing essential information on demographics, disease characteristics, and disease progression. Natural history studies can inform research design by guiding the selection of appropriate inclusion and exclusion criteria, intervention stages, study endpoints, follow-up durations and time points. They can also support the identification and development of biomarkers and may be used as external controls in single-arm trials.
v. Post-Marketing Safety Monitoring and/or Effectiveness Evaluation
Disease registries are characterized by long-term, timely, and longitudinal features. Through long-term post-market monitoring and the continuous accumulation of data, their advantages become more evident. As the number of enrolled patients grows and the observation time extends, registries become increasingly effective at comprehensively monitoring drug safety, detecting rare adverse events, and evaluation of long-term or rare clinical endpoint events that are difficult to obtain in randomized controlled clinical trials. By analyzing registry data, we can not only assess a drug effectiveness in real-world clinical settings, but also support the evaluation of effectiveness under different treatment strategies (such as dosage, treatment duration, frequency of administration, combination therapy, etc.), providing evidence to inform clinical practice and to support label updates or revisions.
vi. Enhancing the Implementation of Clinical Trials
Disease registries can be used in conjunction with the implementation of interventional clinical trials, significantly improving operational efficiency. For instance, in registries with a sufficiently large patient population, screening and enrollment can be expedited by analyzing clinical characteristics and applying the inclusion and exclusion criteria defined in the study protocol. Beyond patient recruitment, randomization can also be integrated into registry-based workflows. A notable example is the SWEDEHEART cardiac registry, which has been used in the development of drugs and medical devices to randomly assign patients within clinical trials. This type of study is known as a registry-based randomized controlled trial (R-RCT).
IV. Establishing Disease Registry Databases
i. Process of Establishing a Disease Registry
The process of establishing a disease registry is illustrated in Figure 1. It begins with the development of an overall plan for the registry, followed by detailed design based on that plan. Next, a database is constructed according to the design requirements. Once the database is established, data collection and entry begin, accompanied by ongoing data quality control. When the database matures, it can be used for analytical purposes. It is also recommended that disease registry databases have the capability for data linkage and expansion. On one hand, this allows for the incorporation of historical data and data from other registries; on the other hand, based on research needs and feasibility of data acquisition, the registry may be expanded in terms of disease types, follow-up durations, variables, and other aspects.
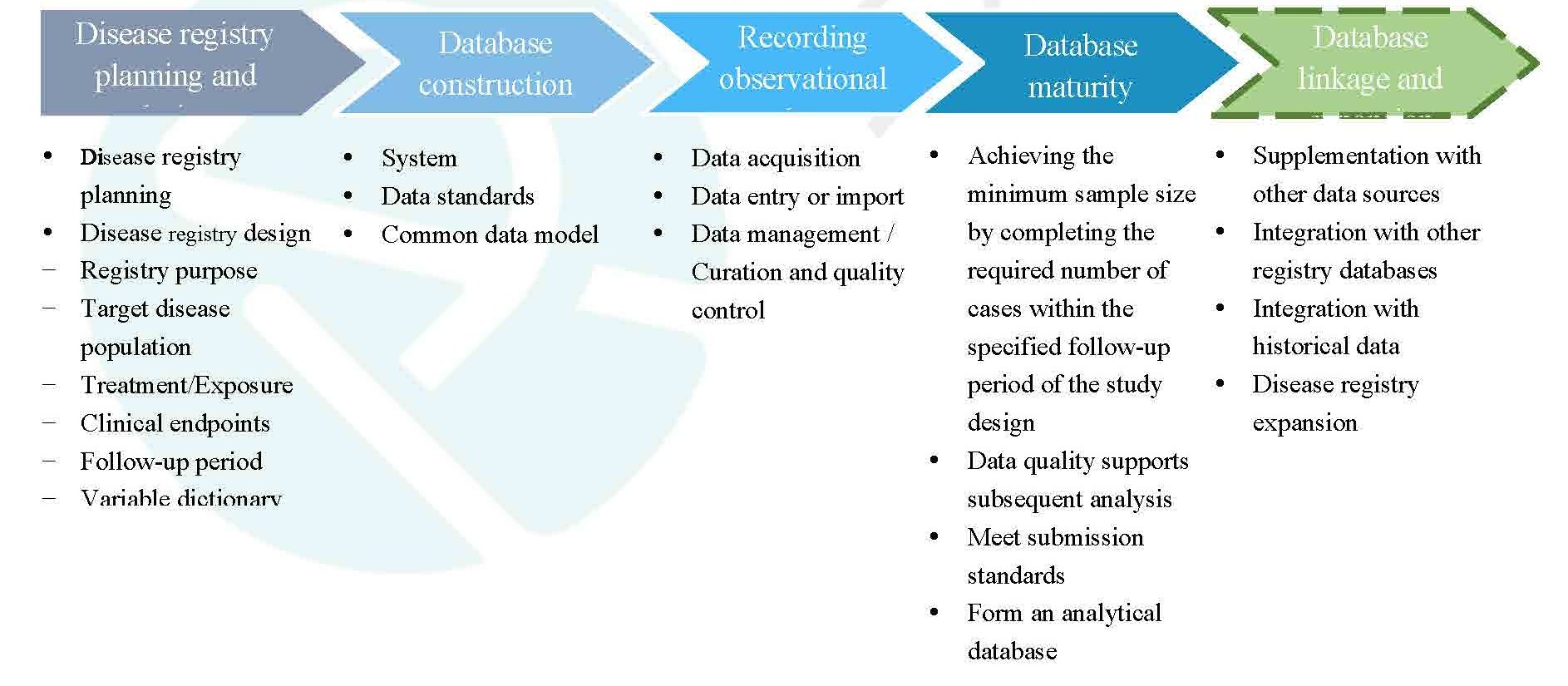
Figure 1. Process of establishing a disease registry
Note: Dashed lines indicate optional steps.
ii. Disease Registry Planning and Design
1. Disease Registry Planning
When developing a disease registry plan, several key considerations should be addressed in sequence, including but not limited to:
Clarifying the purpose of the registry: Clearly define the overall purpose of the registry such as which disease area it targets, the intended scale, and the scope of information to be collected.
Selecting the disease area: This can involve a single disease, a group of related diseases, or multiple disease types. The following sections primarily focus on the single disease registries.
Defining the target disease population: Define the target population based on the selected disease area, and consider the representativeness of the registry population.
Planning the study duration: Determine the follow-up period by balancing scientific considerations, practical feasibility, and funding availability. Clearly define whether the project is intended as a long-term or short-term study. Considering the scientific value and database value, long-term studies are encouraged.
Data sources: Deliberately assess the adequacy, relevance, and accessibility of the data sources.
Data security and ethics: Ensure the security of the data system, compliance in data usage, and address all related ethical considerations.
Identifying stakeholders and collaborators: Disease registries require multidisciplinary collaboration. It is necessary to identify the roles and responsibilities of all involved parties and establish a project team to ensure smooth implementation. Encourage active involvement of patients and collaboration between the project team and patient organizations.
Feasibility assessment: Conduct a comprehensive feasibility assessment to determine whether the project can be successfully implemented. The feasibility assessment should consider various factors including the scientific significance and clinical value of the project, data acquisition and quality assurance, sufficiency of funding, patient compliance, data security and regulatory compliance, ethical risks and other factors.
Project execution plan: It refers to the project’s management and implementation plan, which should include organizing implementation, budget planning and management, progress management, personnel management, quality control, communication mechanisms, and risk management. It should also address data utilization and sharing mechanisms.
2. Disease Registry Design
The Guidance on Real-World Study Design and Protocol Framework (Trial Version) provides detailed discussion on the design of a real-world study. This guidance focuses on several key aspects according to the features of disease registries.
(1) Registry Objectives
A concise description of disease registry should be defined according to the specific disease, target population, relevant treatments, expected outcomes, and primary data sources. The key elements of description include the geographic scope (if applicable), the disease of interest, study duration, target sample size, and the clinical questions designed to be addressed. The clinical questions may be related to, for example, the natural history of rare diseases, evaluation of clinical efficacy, prognostic assessment, safety monitoring, pharmacoeconomic analysis, etc.
(2) Disease Target Population
The disease target population should be clearly defined, including the diagnostic criteria and corresponding disease codes (e.g., ICD codes). Based on target disease, appropriate inclusion and exclusion criteria should be established to determine which population could be enrolled in the registry. To support further evaluation of the representativeness of the study population, key information is recommended to collect such as the details of patient recruitment and study sites, and baseline characteristics.
(3) Treatment/Exposure
In disease registries, treatment strategies and patterns are determined by routine clinical practice. Variables of treatment or exposure should be recorded as comprehensively as possible, such as drug dosage, frequency, route of administration, duration of treatment, combination regimens (if applicable), as well as the brand names and the manufacturers of the drugs. Since established based on the disease, the registries are not typically limited for specific treatments or interventions during development unless for specific considerations. Clinical studies on a specific product or treatment strategy are generally initiated only after assessing the feasibility of the registry database (e.g., identify whether the sample size of the product or treatment strategy is sufficient). Only then, study and control groups are defined and the adjunctive therapies are specified.
(4) Clinical Endpoints
In a clinical study of a specific disease, clinical questions being addressed may vary across studies, and as a result, the primary and secondary endpoints may also differ. Therefore, it often challenges to define primary and secondary endpoints during the initial design of a disease registry. However, it is important to consider what clinical studies the disease registry will support in a long term. These may include primary and secondary endpoints on the effectiveness, safety, and pharmacoeconomic endpoints (if necessary).
To ensure the reliability of clinical endpoint data, it is recommended to incorporate appropriate measures of quality control, such as validation of key data elements of composite endpoints and adjudication of critical clinical events.
(5) Follow-up Period
For short-term studies, the follow-up period is typically determined based on the features of the disease and the key clinical endpoints. For long-term studies, the lower limit of the follow-up period should be specified, while there is generally no requirement for the upper limit.
(6) Variable Dictionary
A dedicated variable dictionary should be developed for a well-standardized disease registry. This dictionary is established upon data elements and primarily includes variable definitions, variable dimensions, variable names and labels, variable types, units of variables, measurement, association of variables, value ranges (e.g., reference ranges), classification and transformation rules, follow-up time points, etc.
Definitions/standard terminology: Each variable should have a corresponding standardized definition. If originated from another terminology library, the definition should be annotated clearly for the source.
Variable dimensions: In a variable dictionary, variables are typically categorized under several dimensions, such as demographics, medical history, diagnosis, treatment/exposure, efficacy outcomes (including primary and secondary endpoints), laboratory tests, safety events, lifestyle and diet, social and environmental factors. Variables should be assigned to appropriate dimensions in a logical manner.
Variable names and labels: Variable names and labels should be defined according to a consistent standard. The maximum length should be noted when variable names set.
Variable types: It is recommended to clearly define the format of data collection for each variable in the variable dictionary. When determining variable types, standardized formats should be used whenever possible such as numeric or option-based (categorical) types. The use of free-text entries is discouraged. For categorical variables, option labels should be encoded in a standardized manner. Uniform coding principles and ordering should be applied throughout the same disease registry.
Association of variables: It is recommended to define the association between variables, such as the association between disease diagnoses, and test results or treatments.
Value ranges: Define the legal value range of variable collection, also known as the valid range. Note that the valid range of a variable refers to whether the variable value is reasonable and valid in the context of the disease registry and it should be distinguished from the clinical normal/reference range of testing result. Reference ranges of laboratory testing results may be updated periodically during the study, and should be managed separately to avoid frequent change in the variable dictionary.
Derived data: Define the methods for generating derived data, such as age groups variables derived from age and body mass index (BMI) derived from height and weight.
Follow-up time points: Each variable should specify its baseline and follow-up features. For variables collected during follow-up, the follow-up intervals and time points may differ from each variable. It is necessary to specify the follow-up period and time window for each variable.
(7) Number of Cases
Generally, during the design phase of a disease registry, it is necessary to estimate the sample size based on one or more specific research questions that need to be addressed most urgently, and to ensure that the final number of cases completing the follow-up period specified in the design is not less than the estimated sample size. In practice, the minimum sample size should be guaranteed while there is no requirement for the upper limit typically. Also, the number of cases will continue to increase as the registry data accumulate. It should be noted that the initial number of cases may not be adequate to support new clinical research questions proposed during the registry development. Therefore, researchers should assess whether current or expanded registry database can meet the sample size requirement of newly proposed study.
(8) Data Source
A disease registry may include both self-collected data and data integrated from external sources. The data sources can be classified into data collected from medical settings and non-medical settings. In medical settings, data are generated from inpatient, outpatient visits, and diagnostic testing results when patients are hospitalized or receive care during scheduled visits. In non-medical settings, data primarily come from out-of-hospital follow-up, PRO/ePRO, and monitoring devices (e.g., wearable devices).
iii. Database Construction
The construction of a disease registry database must fully consider the requirements for interoperability. This includes ensuring communication, data exchange, and information utilization between different databases and systems, from the perspectives of database system, data standards, and common data models (CDM).
1. System:
To ensure data quality, it is recommended to use a validated Electronic Data Capture (EDC) system for data collection and data management/curation in disease registries. The database system used in a disease registry should meet the general requirements of an EDC system, including: 1) a secure physical and network environment; 2) system stability and data security; 3) User role management and access control; 4) audit trail: once data are entered and saved in the system, all audit trails must be recorded and must not be deleted or modified; 5) standardized operating procedures (SOPs). More detailed requirements can be found in the Technical Guidance on Electronic Data Capture in Clinical Trials. In addition, the system must meet interoperability requirements; that is, data transmission and business information exchange between systems should be achieved by defining the structure and format for data exchange.
When a disease registry database is constructed and configured, it is important to fully consider the need for data exchange between multiple databases or data systems. Based on data structures and attributes of the different databases/systems, data exchange standards should be defined in advance, and data transmission testing should be conducted to ensure the accurate transfer of information.
2. Data Standards
During the development of a disease registry, it may be necessary to integrate clinical data from multiple sources. Standardized data structures and formats are the foundation for data exchange and sharing. To enable database linkage and expansion, improve data interoperability and quality and enhance regulatory review efficiency, it is recommended to prioritize industry wide data standards, such as CDASH (Clinical Data Acquisition Standards Harmonization) when building a disease registry database. From a broader perspective, data standards apply to all aspects of disease registry development including protocol design, data collection, analysis, exchange, submission, and report writing. The construction of the registry database should consider the applicability of relevant standards and the compatibility of data structures. For regulatory submissions, the final submitted data must comply with the requirements outlined in the Guidance on Data Submission for Drug Clinical Trials (Trial Version).
For data modules that are not covered by or not applicable to common data standards, unified standards should be developed at the system level of the disease registry and consistently applied across the same or different disease cohorts.
3. Common Data Model
During the design phase of the disease registry database and data collection plan, a clear definition of the applicable common data model for the disease cohort should be established. The mapping relationships between data sources and the CDM should be specified, including variable definitions, rules for data extraction, transformation, and loading (ETL) from source data, and standards for data conversion across multiple databases. For a detailed introduction to common data models, please refer to the Guidance on Using Real World Data to Generate Real World Evidence (Trial Version).
iv. Data Acquisition, Entry, and Quality Control in Disease Registries
Registry data are primarily generated prospectively within the registry system, but may also be integrated with external data sources, such as historical data or data from other systems. This section focuses on how registry data are generated internally within the system.
1. Data Acquisition
The main sources of data in disease registries include hospital information system data, follow-up data, PRO data, individual daily monitoring data, etc.
(1) Hospital Information System Data: Data from hospital internal systems, especially electronic medical records data, are the primary source of disease registry data. Typically, regardless of whether a patient is hospitalized, key data such as baseline information, diagnosis and treatment details, and various test results are all generated within the hospital information systems.
(2) Follow-up Data: Collecting data through patient follow-up is an important approach to ensuring the longitudinal nature of the dataset. According to the study plan, follow-up may be conducted regularly or irregularly, either through clinic visits or remote communication, to collect information on the patient's disease status, clinical endpoints, treatments, and other information. Follow-up data are typically collected using paper or electronic case report forms (CRFs) but validated technical solutions for direct data import may also be considered.
(3) PRO Data: PRO data can be recorded in either paper or electronic formats, with the latter referred to as electronic patient-reported outcomes (ePRO) which is now more commonly used. ePRO systems can be integrated with electronic medical record systems or EDC systems to create a comprehensive, patient-level data flow. For more detailed information on PRO data, please refer to the Guidance on the Use of Patient-Reported Outcomes in Drug Clinical Development (Trial Version).
(4) Individual Monitoring Data: The use of mobile devices (such as smartphones, wearable devices, ambulatory electrocardiogram (ECG) monitors, etc.) offers significant advantages in terms of convenience and timeliness to collect real-time individual monitoring data. This approach not only expands the methods of data acquisition but also enhances the disease registry database. The adoption of advanced and reliable data collection technologies is strongly encouraged in the design and implementation of disease registries.
2. Data Entry or Import:
Data collection can be conducted through both manual entry and automated import methods. Manually entered data typically include paper or electronic follow-up data, paper-based PRO data, etc., which are entered into the EDC system by qualified and authorized personnel. Before initiating the data entry process, it is essential to develop clear guidelines and detailed instructions based on the disease registry’s plan and design specifications. In addition, training should be provided to relevant staff on data filling and entry procedures.
ePRO data, follow-up data from hospital system, and individual monitoring data from mobile devices can be directly imported into the EDC system at times. Prior to import, the mapping relationships between the source data and the EDC database as well as the data import mechanism should be specified and validated through import testing. All regulations and procedures related to the import process should be thoroughly documented to ensure the transparency of the data processing workflow and the traceability of the data.
3. Data Management/Curation and Quality Control
Retrospectively collected data generally requires data curation, while prospectively collected data requires data management. Key steps in the data management/curation process include, but are not limited to: development of a data management/curation plan, design of CRFs/data collection instruments, design and construction of the database, data collection and entry, data verification and query management, medical coding, data review, database locking, data storage and transmission, quality control, etc. In addition, data curation processes also include personal data protection and data security management, establishment of a common data model, as well as data extraction and transformation. For detailed processes, please refer to the Technical Guidance on Data Management for Drug Clinical Trials and the Guidance on Using Real World Data to Generate Real World Evidence (Trial Version).
Data quality control is essential to ensuring the integrity, accuracy, and transparency of research data. Given the characteristics of disease registry data, the following principles are recommended for effective quality control: 1) establish standard operating procedures (SOPs) for quality control; 2) develop a comprehensive quality control plan that clearly outlines the scope, frequency, procedures, and quality standards of quality control activities; 3) clearly define the time points and implementation sequence for data collection within the disease registry plan; 4) provide training and guidance to relevant personnel on data collection, including data entry rules, applicable data standards and common data models, and the proper methods and requirements for data entry, storage, and transmission; 5) ensure the completeness of documentation for all data processing steps; and 6) take proactive measures to manage loss to follow-up, with the goal of maintaining dropout rates within an acceptable range.
v. Database Maturity
After a disease registry database is established, it gradually matures as data are continuously collected and accumulated. A mature database is one in which the generated data can support statistical analyses to produce real-world evidence for regulatory decision-making. Specifically, a mature database should meet the following four criteria: 1) the number of cases that have completed the follow-up period, as defined in the registry design, has reached the minimum required sample size; 2) the data quality is sufficient to support subsequent analysis, meeting the ALCOA+CCEA principles (ALCOA: attributable, legible, contemporaneous, original, accurate; CCEA: complete, consistent, enduring, available when needed); 3) the data can be converted into formats that meet regulatory submission standards, as specified in the Guidance on Data Submission for Drug Clinical Trials (Trial Version); and 4) the data can be used to generate analysis-ready datasets tailored to diverse research needs.
vi. Database Linkage and Expansion
During the development of a disease registry, in addition to building the database itself, it is also possible to integrate external data sources, including pre-existing databases and other registries. Moreover, the registry can be expanded based on emerging research needs or newly conditions.
1. Database Linkage
If the registry system cannot obtain all necessary information for the registry-based study from its own internal data sources, it is essential to supplement the data with external data sources. For example, in oncology studies where mortality is the primary endpoint, a disease registry relying mainly on hospital information systems may not include complete death records. In such cases, information would need to be integrated from other sources (such as the national CDC death registration system). When integrating with external data sources, it is essential to ensure interoperability between systems and to guarantee that data transmission is accurate, consistent, and complete.
When integrating with other registry databases, differences in database structures and data standards must be considered. This may require the use or development of a common data model and assessment of the feasibility of linkage, including the degree of alignment of key variables, data applicability, and data traceability.
When integrating pre-existing data, it is also necessary to ensure that the merged cases are followed up in accordance with the prospective designed follow-up plan, especially for those whose follow-up duration does not meet the pre-defined requirements. If the quality of the historical data is poor—such as high rates of missing key variables or inaccurate records—or if there are inconsistencies in the definitions and measurements of the target population or key variables compared to the current disease registry, then even if the data are accessible, they should not be integrated.
2. Data Expansion
As scientific research advances and data accessibility improves, disease registries may be expanded in various ways.
· Addition of disease categories: For example, to support comorbidity research, a chronic kidney disease cohort may be added to an existing diabetes registry; or for convenience in managing related diseases, a heart failure cohort may be added to a coronary heart disease registry.
· Extension of cohort follow-up period: For some disease registries with relatively short follow-up periods, the original study plan may be modified to significantly extend the follow-up duration—either due to the limitations of the initial design (e.g., follow-up was too short to estimate median survival time) or improvements in research conditions (e.g., securing adequate funding). A longer follow-up period increases the scientific value of the cohort study.
· Addition of variables: As scientific research advances—bringing new diagnostic methods, prognostic factors (e.g., novel biomarkers), endpoint evaluation methods, and classification criteria—disease registries should promptly incorporate newly relevant variables to keep pace with evolving scientific needs and landscape.
vii. Data Security
Data security in disease registries should be given high priority. Data used for analysis must be anonymized and must not contain any sensitive personal information, such as names, identification numbers, contact details, affiliations, home addresses, office addresses, etc. In both the data management interface and the final processed analysis database, the patient identifier should be a study ID. This ID is typically generated by converting personal information at the point of data generation (e.g., from original medical records or HIS). If an existing registry has not undergone anonymization, it must be properly anonymized before it can be shared; otherwise, it may raise legal concerns regarding personal information security. For specific measures on data security, please refer to the Guidance on Using Real World Data to Generate Real World Evidence (Trial Version).
viii. Early Integration of PFDD Concept
Patient involvement in disease registries offers substantial value. Engaging patients in defining study endpoints, key variables, and evaluation criteria helps ensure that the research reflects patient priorities—such as quality of life, treatment satisfaction, and adherence. Incorporating disease management functionalities into registry design—such as online medication and monitoring guidance, psychological support, and rehabilitation training—can empower patients to better manage their condition and actively benefit from the drug development process. Aligned with the PFDD approach, disease registry systems can also serve as platforms to provide patients with information about their condition, available treatment options, and opportunities to participate in research, thereby improving disease awareness and health literacy. Furthermore, sharing registry-related information—either through patient organizations or directly with patients—facilitates broader recruitment, improves study population representativeness, and reduces selection bias.
Overall, integrating the PFDD concept into disease registries and related research contributes to greater patient satisfaction, optimizes clinical practice, and improves the quality and efficiency of drug development.
V. Evaluation of Disease Registry Data from a Clinical Research Perspective
Only real-world data that have been properly assessed for applicability can potentially generate real-world evidence that supports regulatory decision-making. The evaluation of disease registry data applicability can refer to the Guidance on Using Real World Data to Generate Real World Evidence (Trial Version). The following section outlines key considerations for evaluating disease registry data.
i. Achievement of Research Objectives
The first step is to evaluate whether the disease registry data can support the intended research objectives—that is, whether the RWE generated through analysis of these data can adequately address the clinical or scientific questions of the study. A preliminary assessment should include: reliability of the diagnostic methods for the disease; alignment of case selection with the study objectives; Completeness of treatment details for study and control groups (if applicable); credibility of the measurement of key clinical outcomes; adequacy of follow-up duration for key outcomes; integrity of key recorded variables; sample size adequacy for statistical requirements; documentation of critical safety information; implementation of effective quality assurance measures in place; data accessibility and traceability.
ii. Standardization of Disease Registry Development
The standardized development of disease registries is critical to ensure the scientific integrity and operational efficiency of research. Standardization should be evident in a well-defined plan and design, followed by rigorous implementation and robust quality control. Key aspects to evaluate also include: adoption of industry-wide standards; consistent application of uniform protocols across the cohort; availability of a dedicated variable dictionary, operations manual, and SOPs; implementation of informed consent procedures and patient privacy protections; assurance of data security; comprehensive documentation of operational processes; clearly defined roles and access permissions for data management and usage; and effective organizational structure and communication mechanisms.
iii. Representativeness of Registry Population
The representativeness of the registry population to the target population should be evaluated based on the inclusion and exclusion criteria defined in the study protocol. Substantial differences between the two populations may introduce potential biases or limit the generalizability of the study findings. In cases where data are integrated from multiple sources, the consistency of the study population should also be assessed, for example, by comparing baseline characteristics.
iv. Follow-up Duration, Follow-up Interval, and Time Window
In a disease registry, the follow-up period must be sufficiently long to capture key outcome variables; otherwise, accurate and reliable estimations cannot be made. For example, if the primary endpoint of a study is survival time, a short follow-up period that does not capture the median time to event would make it difficult to objectively assess treatment effectiveness. On the other hand, if the outcome of interest requires an especially long follow-up period—such as in chronic lymphocytic leukemia, where survival times are long and highly variable—a follow-up period of 20 years or more may be necessary. In such cases, overly ambitious follow-up may be impractical without stable funding and dedicated teams.
The design of follow-up intervals should be reasonable. If the intervals are too long, it may hinder the understanding of disease progression patterns and reduce the accuracy of estimating time-dependent events. Conversely, excessively short intervals can increase the burden of research implementation and the operational complexity, potentially affecting the overall quality of the study. Although time windows in disease registries can be more flexible than in randomized controlled trials, they should still be appropriately defined to avoid compromising the quality of the research.
v. Data Quality and Control
The evaluation of data quality is thoroughly addressed in the Guidance on Using Real World Data to Generate Real World Evidence (Trial Version). Given the long-term nature of disease registries, the daily operation and maintenance of the registry are especially important. As such, special attention should be given to the quality control plan and its implementation. Key considerations include whether a detailed quality control plan is in place, whether dedicated quality control personnel are assigned, whether appropriate SOPs are established, and whether there is a systematic training program with corresponding documentation of activities. In addition, data traceability is especially critical in disease registries; for any integrated external data, traceability must also be fully ensured.
VI. Submission of Disease Registry Data and Communication with Regulatory Authorities
i. Submission of Disease Registry Data
Disease registry data intended to support regulatory submissions must comply with the requirements outlined in the Guidance on Using Real World Data to Generate Real World Evidence (Trial Version) and the Guidance on Data Submission for Drug Clinical Trials (Trial Version).
ii. Communication with Regulatory Authorities
To ensure that disease registry data meet regulatory quality requirements, it is essential to engage in timely communication with the Center for Drug Evaluation when real-world research using registry data is intended as pivotal evidence to support regulatory approval. Before formally initiating a real-world study, discussions should address the study objectives, relevance, and whether the registry data satisfy regulatory requirements for the applicability of RWD. Key discussion topics include the planning and design of the registry, its operational plans and maintenance protocols, the sample size and duration of the cohort, key variables, data completeness, and the proposed data curation strategy and plan. For specific timing and detailed considerations regarding such communication, refer to the Guidance on Communication of Real-World Evidence to Support Drug Registration Applications (Trial Version).
References
[1] National Medical Products Administration. Guidance on Using Real-World Evidence to Support Drug Development and Regulatory Review (Trial Version). 2020.01
[2] National Medical Products Administration. Guidance on Using Real World Data to Generate Real World Evidence (Trial Version). 2021.04
[3] National Medical Products Administration. Guidance on the Use of Patient-Reported Outcomes in Clinical Development of Drugs (Trial Version). 2021.12
[4] AHRQ. Registries for Evaluating Patient Outcomes A User's Guide. 4th Edition. 2020.09
[5] EMA. Guideline on Registry-based Studies. 2021.12
[6] FDA. Real-World Data: Assessing Registries to Support Regulatory Decision-Making for Drug and Biological Products. 2023.12
[7] National Medical Products Administration. Technical Guidance on Real-World Study Supporting Research & Development and Evaluation of Pediatric Drugs (Trial Version). 2020.08
[8] National Medical Products Administration. Technical Guidance on Clinical Development of Drugs for Rare Diseases (Trial Version). 2022.01
[9] National Medical Products Administration. Guidance on Statistical Considerations for Clinical Research on Drugs for Rare Diseases (Trial Version). 2022.06
[10] National Medical Products Administration. Technical Guidelines on the Applicability of Single Arm Clinical Trials Design to Support Marketing Authorization Applications of Antitumor Drugs (Trial Version). 2023.03
[11] FDA. Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products (Draft). 2023.02
[12] National Medical Products Administration. Guidance on Real-World Study Design and Protocol Framework (Trial Version). 2023.02
[13] National Medical Products Administration. Technical Guidance on Electronic Data Capture in Clinical Trials. 2016.07
[14] National Medical Products Administration. Guidance on Data Submission for Drug Clinical Trials (Trial Version). 2020.07
[15] National Medical Products Administration. Technical Guidance on Data Management for Drug Clinical Trials. 2016.07
[16] National Medical Products Administration. Guidance on Communication of Real-World Evidence to Support Drug Registration Applications (Trial Version). 2023.02
Product Registry: A registry focused on one or more specific products.
Single-arm/One-arm Trial: A non-randomized clinical trial with only an experimental group, typically using an external control, such as a historical or concurrent control.
Registry: A database created by the organized, systematic, and standardized collection of longitudinal, patient-level data on demographics, disease, exposures, diagnosis and treatment, and outcomes, based on predefined objectives and follow-up/observation timelines.
Registry-based Study: A study conducted using data generated from a registry.
Electronic Medical Record, EMR: An electronic record of health-related information for individual patient, which is created, collected, managed, and accessed by authorized clinical professionals within a healthcare institution.
Observational Study: A study with specific research questions that investigates the causal relationship between exposure/treatment and outcomes without active intervention in natural or clinical populations.
Patient-reported Outcome, PRO: An outcome assessment reported directly by the patient that reflects their experience of a disease and treatment, without interpretation or modification by others.
Patient-focused Drug Development, PFDD:
A systematic approach to help ensure that patients’ experiences, perspectives, needs, and priorities are captured and meaningfully incorporated into the development and evaluation of medical products throughout the medical product life cycle.
Patient Experience Data, PED/ Patient Input: Any information voluntarily provided by individuals on patients’ experience with a disease or condition. Such information includes patients’ experiences, perspectives, needs, and preferences, but is not limited to symptoms and the natural history of the disease, the impact of the condition on function and quality of life, treatment experiences, the importance of outcomes to patients, patient preferences for outcomes and treatments, and other information that is important to patients.
Retrospective Observational Study: An observational study in which the target population is defined at study’s initiation, and the research is conducted using historical data (i.e., data generated before the study began).
Clinical Trial: A type of interventional clinical study in which one or more interventions (which may include placebos or other controls) are prospectively assigned to human participants to evaluate the effects on health-related biomedical or behavioral outcomes.
Disease Registry: A registry focused one specific disease, a group of related diseases, or multiple disease types.
Prospective Observational Study: An observational study that predefines data collection for exposure/treatment and outcome in the target population at the start of the study.
Data Standard: A set of rules for how a specific type of data is structured, defined, formatted, or exchanged across computer systems. Data standards help to ensure that submitted data are predictable and consistent, and compatible with information technology systems or scientific tools.
Data Linkage: The process of combining, connecting, and integrating data and information from multiple sources to create a unified dataset.
Data Element: A single observation recorded about a study subject in clinical research, such as date of birth, white blood cell count, pain severity, or other clinical observations.
Data Curation: The process of preparing raw data for statistical analysis to address a specific clinical research question. It includes, at minimum: data extraction (including from multiple sources), data security handling, data cleaning (logical checks, outlier handling, ensuring completeness), Data transformation (e.g., common data model, normalization, natural language processing, medical coding, derivation of variables), data quality control, data transfer and storage, etc.
Common Data Model, CDM: A data system developed through multidisciplinary collaboration to enable the rapid integration and standardization of heterogeneous data from multiple sources. Its primary function is to convert source data with different standards into a unified structure, format, and terminology, facilitating data linkage across databases or datasets.
External Control: In clinical trials, an external control uses data not derived from the trial subjects to evaluate the effects of the intervention under study. External controls may come from historical data, concurrent observational data, or target values.
Source Data: All information recorded in clinical research that relates to clinical symptoms, observations, and other activities necessary for reconstructing and evaluating the study, documented in original records or certified copies. Source data are contained in source documents (including the original records or their verified copies).
Real-World Data, RWD: Data related to patient health status and/or diagnosis, treatment, and healthcare, routinely collected from daily settings. Not all real-world data can generate real-world evidence—only RWD that meets criteria for relevance and appropriateness can potentially produce real-world evidence.
Real-World Research/Study, RWR/RWS: A research process designed to answer predefined clinical questions by collecting (real-world) data related to patient health status and/or diagnosis, treatment, and healthcare of study subjects in real-world settings, or by using summary data derived from such sources. Through analysis, it aims to obtain clinical evidence (real-world evidence) regarding drug use and its potential benefit-risk profile. The main study type is observational study, though pragmatic clinical trials may also be included.
Real-World Evidence, RWE: Clinical evidence about a drug’s usage and potential benefit-risk profile, derived from appropriate and rigorous analysis of applicable real-world data.
Appendix 2. Chinese and English Terminology
|
中文 |
英文 |
|
标准操作规程 |
Standard Operating Procedure, SOP |
|
病例登记 |
Patient Registry |
|
单臂临床试验 |
Single-arm/One-arm Trial |
|
登记 |
Registry |
|
登记研究 |
Registry-based Study |
|
电子病历 |
Electronic Medical Record, EMR |
|
观察性研究 |
Observational Study |
|
患者报告结局 |
Patient Reported Outcome, PRO |
|
患者为中心的药物研发 |
Patient-focused Drug Development, PFDD |
|
患者体验数据 |
Patient Experience Data/ Patient Input |
|
回顾性观察性研究 |
Retrospective Observational Study |
|
疾病登记 |
Disease Registry |
|
基于登记的随机对照临床试验 |
Registry-based/Register-based RCT |
|
前瞻性观察性研究 |
Prospective Observational Study |
|
数据标准 |
Data Standard |
|
数据融合 |
Data Linkage |
|
数据元素 |
Data Element |
|
数据治理 |
Data Curation |
|
通用数据模型 |
Common Data Model, CDM |
|
外部对照 |
External Control |
|
源数据 |
Source Data |
|
卫生信息系统 |
Health Information System, HIS |
|
健康服务登记 |
Health Service Registries |
|
医疗产品登记 |
Product Registry |
|
真实世界数据 |
Real World Data, RWD |
|
真实世界研究 |
Real World Research/Study, RWR/RWS |
|
真实世界证据 |
Real World Evidence, RWE |
|
质量控制 |
Quality Control, QC |
|
药物注册 |
Drug Registration |
|
可归因性 |
Attributable |
|
易读性 |
Legible |
|
同时性 |
Contemporaneous |
|
原始性 |
Original |
|
准确性 |
Accurate |
|
完整性 |
Complete |
|
一致性 |
Consistent |
|
持久性 |
Enduring |
|
可获得性 |
Available When Needed |
点击此处,查看原文附件
Guidance on the Application of Real-World Data Based on Disease Registries (Final)
- Guidance on the Application of Real-World Data Based on Disease Registries (Final)04-28
- Guidance on the Design and Protocol Development of Real-World Studies for Drugs (Final)03-20
- Guideline on Using Patient Reported Outcomes in Drug Clinical trail (Final)03-17
- Guidance on Communications of Using Real-World Evidence to Support Registration Applications for Drug and Biological Products (Final)03-17
- Guideline on Using Real-World Study to Support the Development and Evaluation of Pediatric Drugs (Final)03-17